Our laboratory specializes in research that might yield breakthroughs in clinical medicine based on concepts derived from microscopy observations of biopsy or surgical specimens for pathological diagnosis. Our plan is to collect high-quality tissue specimens of cancers from various organs that we ourselves have pathologically diagnosed, and to develop a strategy for multilayered omics analysis focusing mainly on the epigenome. In this way, we hope to clarify the molecular mechanisms of cancer and cancer-related diseases to advance understanding of how the biological characteristics of cancer are determined. The aim of our studies is to develop strategies for carcinogenetic risk estimation, early diagnosis of cancer, prognostication and companion diagnosis, and to identify candidate molecules for cancer prevention and therapy.
This laboratory used to be located in the Pathology Division, National Cancer Center Research Institute (Division of Molecular Pathology after reorganization) until June 2015, and moved to the Department of Pathology, Keio University School of Medicine, on July 1, 2015. Along with this move, our laboratory was re-established at the Shinanomachi Campus East School Building and Museum. At the National Cancer Center Research Institute, we were engaged in mission-oriented cancer research aimed at “significant reduction in cancer morbidity and mortality”, and performed nationwide joint research projects with private companies and other universities and research institutes. Based on this experience, we are now working on industry-academia-government collaborative research, which is important for realizing our goal of multilayer/integrated omics analysis, in addition to our own specialized epigenomic analysis, and finishing the results in a form that can actually be delivered to patients.
We have vigorously worked towards clarifying the significance of epigenome abnormalities during multistage human carcinogenesis (especially the precancerous stage). Around 1995, when many researchers understood that “cancer is a disease of the gene” (caused by gene mutations), Kanai and co-workers considered that the diversity of cancers, in terms of the histological heterogeneity observed in daily microscope work, could not be fully explained on the basis of a genetic mechanism (i.e. irreversible gene mutation). Therefore, although most researchers at the time had ignored the involvement of epigenetics in carcinogenesis, Kanai’s group started to focus on this issue (particularly DNA methylation abnormalities), ahead of the global research trend.
At a time when only the Rb and VHL tumor suppressor genes were known to be silenced by DNA methylation, we reported that the CDH1 tumor suppressor gene in human cancers is inactivated by two hits: DNA hypermethylation and loss of heterozygosity (LOH) (Proc Natl Acad Sci USA, 92: 7416, 1995). Around the same time, we reported that DNA methylation alteration had already occurred frequently in liver tissue showing chronic hepatitis and cirrhosis, which are widely considered to represent a precancerous stage for hepatocellular carcinoma, from patients with hepatitis virus infection (Jpn J Cancer Res, 87: 1210, 1996). This was one of the earliest reports of DNA methylation abnormalities at the precancerous stage. Since many researchers at the time expected that there should be no molecular abnormalities in non-cancerous tissue, they dismissed as artifacts slight bands indicating DNA methylation abnormalities that were evident on Southern blotting of samples derived from non-cancerous tissue obtained from cancer patients. In order to strengthen our case, we have been accumulating data on methylation alterations of genomic DNA extracted from pathological specimens reflecting various steps of multistage carcinogenesis in various organs, using appropriate analytical techniques.
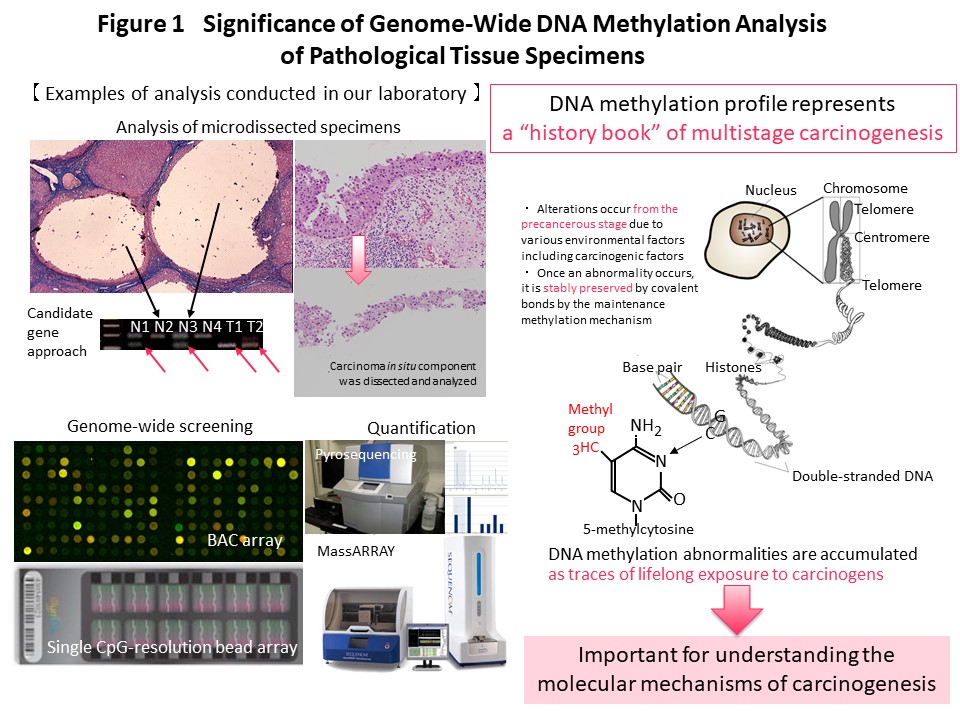
Recently we have performed genome-wide DNA methylation (methylome) analysis and clearly demonstrated that DNA methylation alterations occur frequently in precancerous conditions and precancerous lesions of various organs with background factors such as chronic inflammation accompanied by persistent infection such as viruses, and also smoking. It was shown that DNA methylation abnormalities that occurred at the precancerous stage were inherited by the cancers themselves and defined their clinicopathological aggressiveness and the prognosis of patients. DNA methylation abnormalities at the precancerous stage are maintained by a mechanism involving the DNA methyltransferase DNMT1, and accumulate in the genome, representing a “record” of lifetime exposure to carcinogenic factors. Thus, in each individual patient, the DNA methylation profile can be considered to represent the “history” of multistage carcinogenesis. We are continuing to investigate cancer epigenomics because we consider it to be particularly important for understanding the molecular basis of carcinogenesis (Fig. 1).
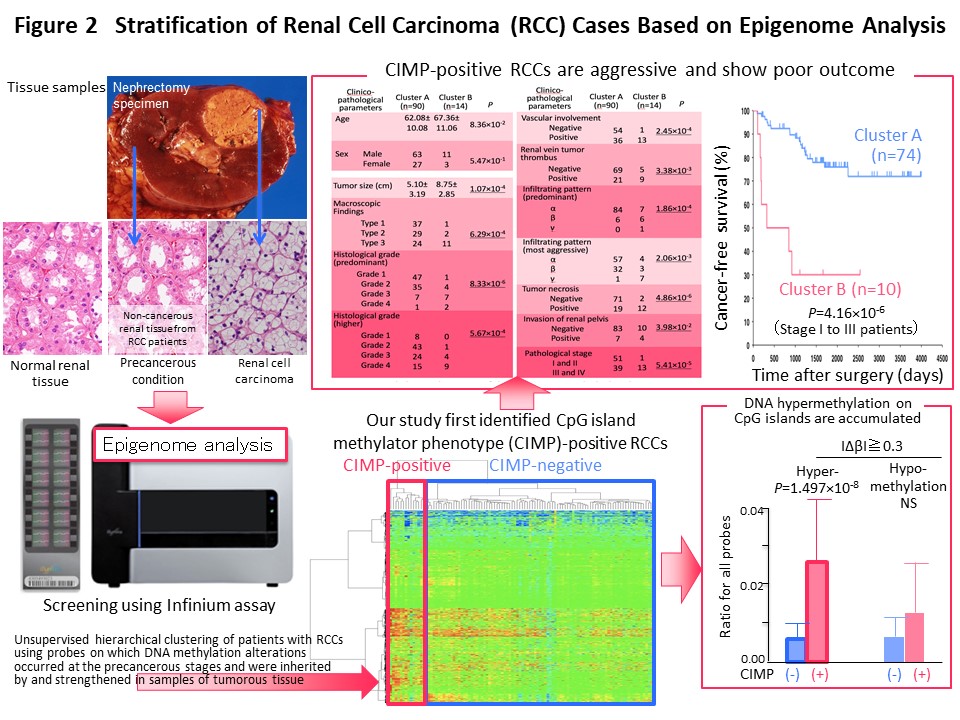
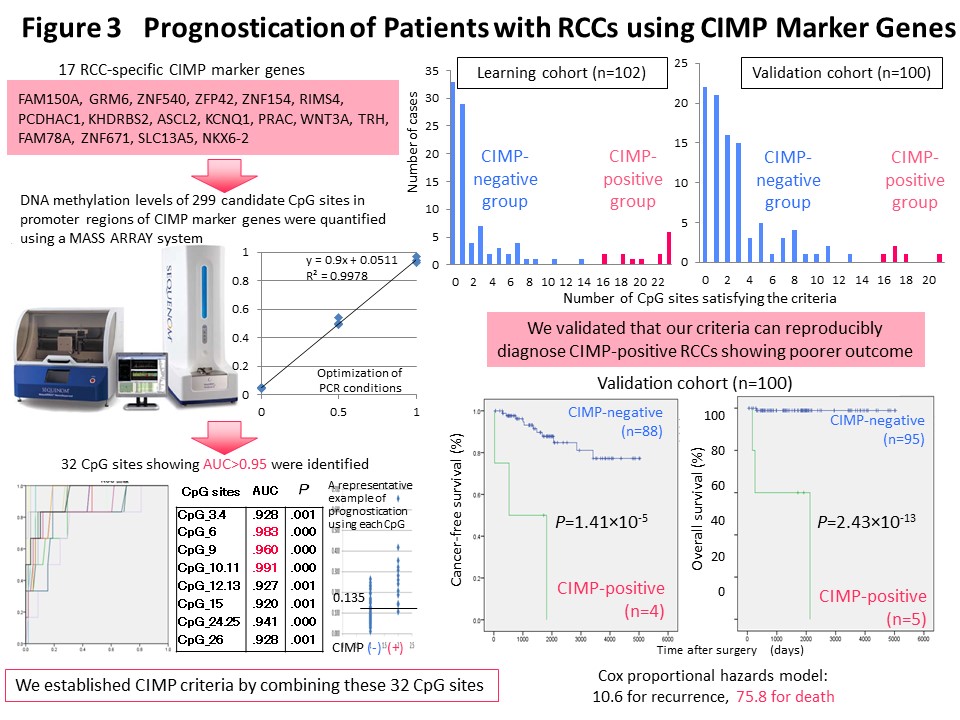
We think that it makes sense to stratify cancer cases based on their epigenomic profile, which represents a “record” of the various stages of carcinogenesis, and to integrate the results of multilayered omics analyses including the genome, epigenome, transcriptome, proteome and metabolome, for each stratified cluster of cancer cases in order to explore the molecular mechanisms of carcinogenesis. With this in mind, Kanai has conducted a joint research project aimed at “comprehensive discovery of drug targets based on multilayer disease omics analysis” (National Institute of Medical Innovation) (2010-2014) as the principal investigator in the epigenome analysis team. Among renal cell carcinomas, those with positivity for the CpG island methylator phenotype (CIMP) and a poor prognosis were identified on the basis of the epigenomic profile, which is altered even from the precancerous stage (Fig. 2) (Carcinogenesis, 33: 1487, 2012). We have developed CIMP-based diagnostic criteria for reproducible diagnosis of such CIMP-positive cases by identifying renal cell carcinoma-specific CIMP marker genes and quantifying their DNA methylation levels (Fig. 3) (BMC Cancer, 14: 772, 2014). Furthermore, multilayer/integrated omics analysis showed that Aurora kinases are a therapeutic target for CIMP-positive cases (Fig. 4) (Int J Cancer, 137: 2589, 2015). The results of this project are deposited in the “Integrative Disease Omics Database (iDOx DB)” (https://gemdbj.ncc.go.jp/omics/docs/disease.html).
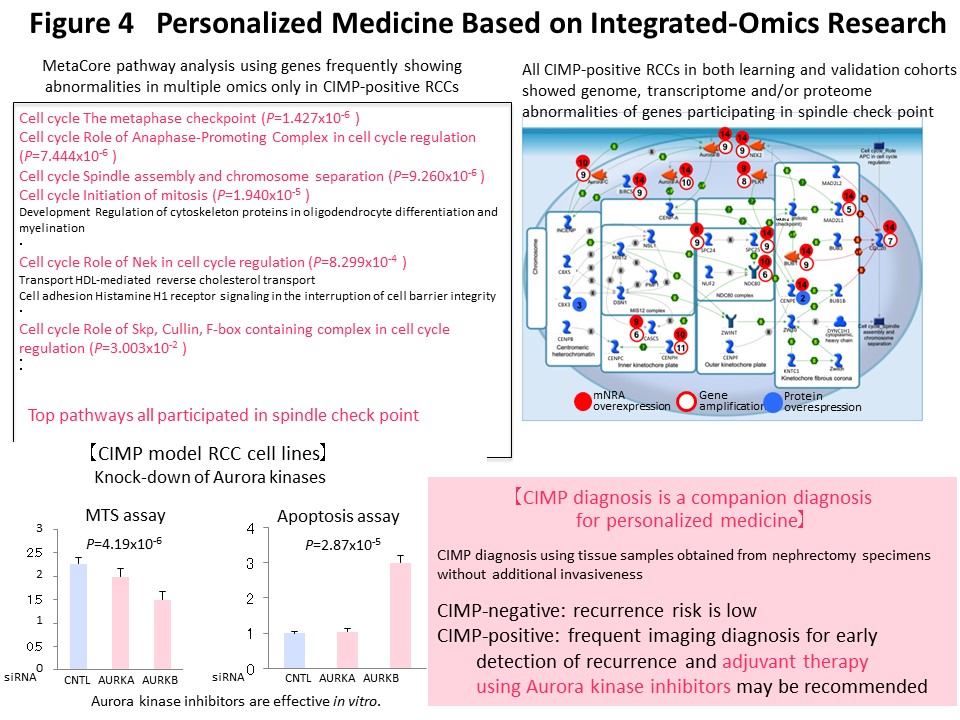
This multilayered disease omics analysis was inherited as a GAPFREE (Funding for Research to Expedite Effective drug discovery by Government, Academia and Private partnership) project in which Kanai has continuously participated as the principal investigator in the epigenome analysis team. Through this collaborative research, we have been providing epigenome data for identification of drug targets for cancer, chronic kidney disease, and neurologic disorders.
For the success of omics analysis such as genomic research using pathological tissue specimens, it is important to use high-quality specimens collected appropriately. In this context, to facilitate collection of a sufficient number of high-quality samples suitable for research at universities, hospitals and biobanks, the Japanese Society of Pathology (JSP) has established “Guidelines on the handling of pathological tissue samples for genomic research”, which define the appropriate operating procedures for collection, storage, and transfer of tissue samples, with the support of the Ministry of Education, Culture, Sports, Science and Technology and the Japan Agency for Medical Research and Development. Kanai was chairman of the Committee for establishment and dissemination of the Guidelines, and worked with researchers who are members of the committee. In particular, Parts 2 and 3 of these guidelines were established from abundant data that had been empirically analyzed after storage and preparation of pathological tissue samples using various methods and conditions.
The Guidelines in printed booklet form have been shipped nationwide, and a full text version is available on the JSP web page (http://pathology.or.jp/genome/), which also includes an e-learning system (http://pathology.or.jp/genome/e-Learning/). We have also explained the Guidelines at workshops held by the JSP (Fig. 5). The Guidelines have been edited so that they constitute “rules with a scientific basis”, the text being arranged side by side with the empirical data (Fig. 6). The English version of the Guidelines was published in Pathology International (68: 63, 2018) (Fig. 7). In the future, we will continue to revise the Guidelines by adding empirical data to support newly devised techniques. We hope that pathologists, clinicians, clinical laboratory technicians, biobank practitioners, and genomic researchers at universities and hospitals throughout Japan will become familiar with the appropriate procedures.



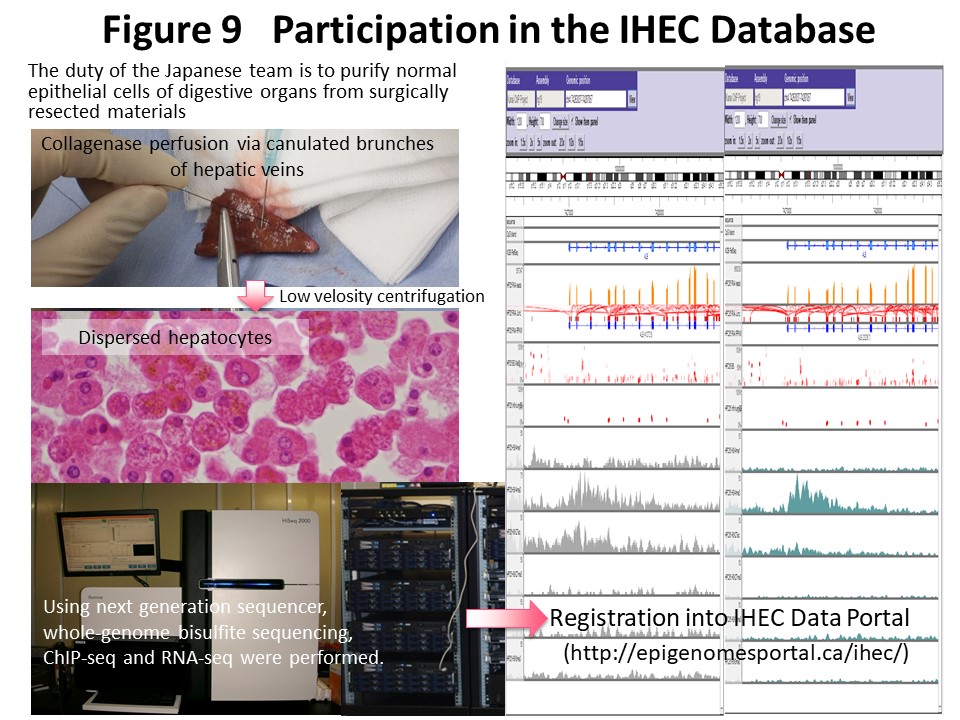
Epigenome information consisting mainly of DNA methylation and histone modification patterns shows diversity among tissues or cell lineages and also heterogeneity based on differences in race and environmental factors. The IHEC (http://www.ihec-epigenomes.org/) was established to gain an understanding of epigenome diversity and build an epigenome database as a common platform for research worldwide. Our laboratory is supported by the Japan Agency for Medical Research and Development CREST research project and participates in the IHEC as a representative research team in Japan (Fig. 8). Whole-genome bisulfite sequencing, chromatin immunoprecipitation (ChIP) sequencing, RNA sequencing and whole-genome sequencing have been performed using a next-generation sequencer for normal epithelial cells of the liver, colon, stomach, and kidney obtained at surgery from Japanese individuals in collaboration with researchers at the National Cancer Center, Kyushu University, and the University of Tokyo (Fig. 9). We have disclosed the results through the National Bioscience Database Center (NBDC) (https://humandbs.biosciencedbc.jp) and the IHEC Data Portal (http://epigenomesportal.ca/ihec/) and made international contributions. Kanai is a member of the IHEC Bioethics Committee, and the “IHEC Science Day & Annual Meeting 2015” was co-sponsored by the CREST-IHEC Kanai Research Team and the Japan Agency for Medical Research and Development in Tokyo (Fig. 10).


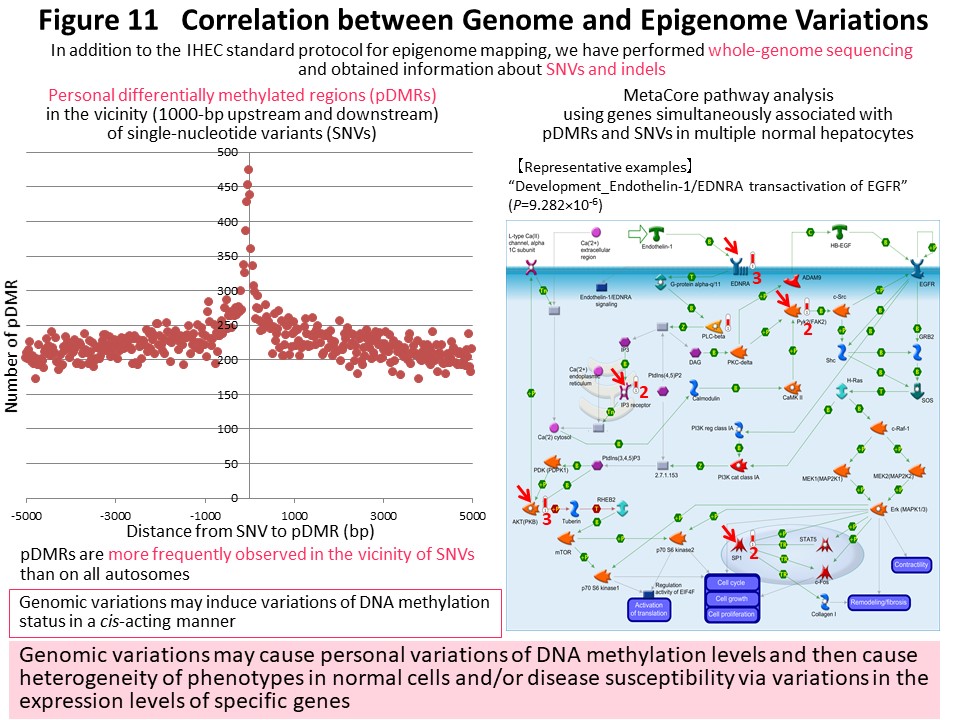
Based on data acquired by the IHEC, we have identified genomic regions where personal differences in DNA methylation levels exist among healthy Japanese individuals (personal differentially methylated regions [pDMRs]). pDMRs were frequently observed in the vicinity of polymorphic sites showing single-nucleotide variation or insertion and deletion in these individuals. We believe that genomic polymorphisms induce epigenomic polymorphism (pDMRs) in a cis-acting manner, thus creating individual differences in phenotype and disease susceptibility through individual differences in gene expression (Fig. 11) (Epigenomics, 10: 955, 2018). By comparison with standard epigenomic profiles of normal cells registered in the IHEC database, we would like to identify disease-specific epigenomic profiles for further clarification of the molecular mechanisms of carcinogenesis and development of biomarkers and drug targets.
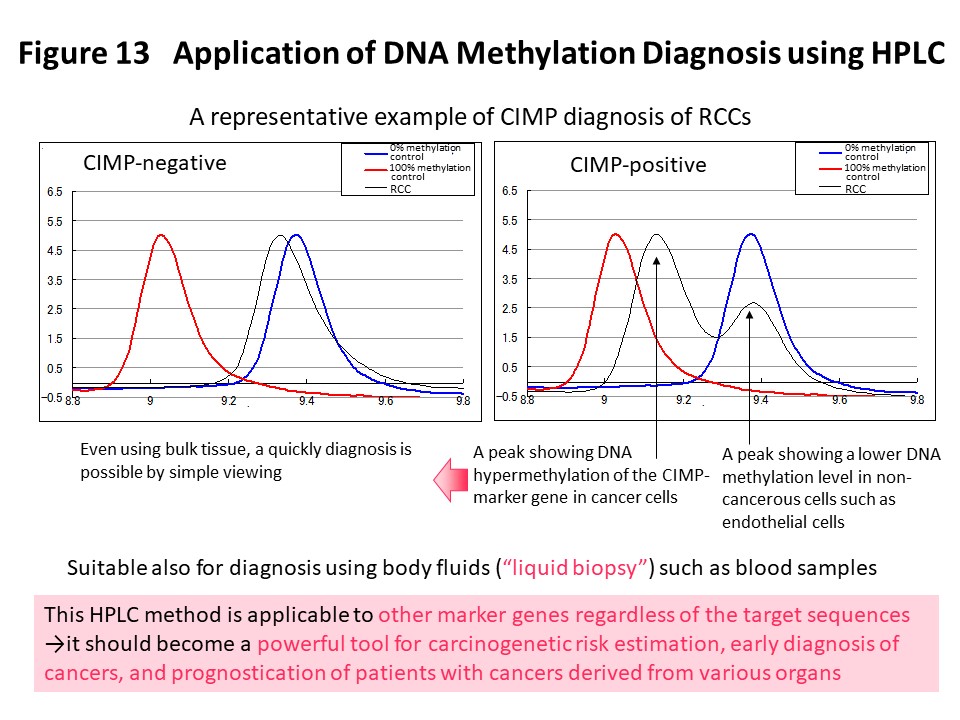
Using the DNA methylation levels of the renal cell carcinoma-specific CIMP marker genes in Fig. 3, it may be possible to implement prognostication and companion diagnosis for postoperative adjuvant therapy (Fig. 4) in patients with CIMP-positive renal cell carcinoma. In order to put such DNA methylation diagnostics into practical use, we and Sekisui Medical Co., Ltd. have newly developed a DNA methylation diagnostic system based on HPLC that is compact and easy to operate, highly quantitative, and can be introduced into the laboratory of any hospital (Cancer Sci, 109: 1690, 2018). A press release has been issued (https://www.ncc.go.jp/jp/information/pr_release/2015/0317/index.html), and reported in several media including national newspapers (Fig. 12). Domestic and international patents have also been granted (Fig. 12). The HPLC column we have developed contains a packing material consisting of ammonium cations and hydrophobic particles. Since thymine, which is equivalent to unmethylated cytosine after bisulfite conversion, interacts more strongly with the column packing material, the elution time is delayed and the DNA methylation levels can be accurately quantified (Fig. 12). Fig. 13 shows an example of CIMP diagnosis of renal cell carcinoma using this principle. Even if bulk tissue is used, it is easy to obtain a chromatogram in a short time with a peak for vascular endothelium showing a low level of DNA methylation and a population of cancer cells showing DNA hypermethylation of the CIMP marker gene. In addition to CIMP diagnosis of renal cell carcinoma, we intend to apply our new diagnostic system to carcinogenetic risk estimation, early diagnosis, prognostication, and companion diagnostics for molecular-targeted therapy using specimens of not only tissue, but also blood and urine (so-called liquid biopsy) for patients with cancers derived from various organs.

In order to obtain clinically useful research results, we continuously conduct genome-wide DNA methylation analysis of large numbers of pathological tissue specimens showing various clinicopathological features from patients with cancers derived from various organs. Recently we have been employing an excellent screening method using a high-density bead array, which is applicable to a large number of specimens and yields a sufficient amount of information. We are especially interested in the significance of DNA methylation alterations at the precancerous stage in various organs. For example, gastric mucosa with chronic gastritis or Helicobacter pylori infection may be at the precancerous stage for gastric cancer. Stratification of gastric cancer cases based on the DNA methylation profiling of the non-cancerous gastric mucosa, rather than the cancerous tissue itself, was significantly correlated with clinicopathological aggressiveness and poorer patient outcome (Carcinogenesis, 36: 509, 2015). We believe that the DNA methylation profile established at the precancerous stage is inherited by cancer and determines the biological characteristics of the latter. Non-cancerous lung tissue obtained from patients with lung adenocarcinoma also had a DNA methylation profile that responded to background factors of carcinogenesis such as smoking and chronic obstructive pulmonary disease (Int J Cancer, 135: 319 , 2014). Surprisingly, it was found that the DNA methylation profile at the precancerous stage (non-cancerous lung tissue) was more likely to determine the prognosis than the DNA methylation profile of the lung adenocarcinoma tissue itself (PLoS One, 8: e59444, 2013). Urothelial carcinoma is clinically noteworthy for its multicentricity and tendency for recurrence: synchronously or metachronously multifocal urothelial carcinomas often develop in the renal pelvis, ureter and urinary bladder. This is because the urothelium of the entire urinary tract has been continuously exposed to carcinogens in the urine and is at the precancerous stage. In order to prevent the occurrence of urothelial carcinoma and enable early diagnosis, we have developed carcinogenetic risk estimation criteria that may allow detection of precancerous DNA methylation abnormalities in urine samples. In addition, we are developing prognostic criteria for prediction of early recurrence of various organ cancers.
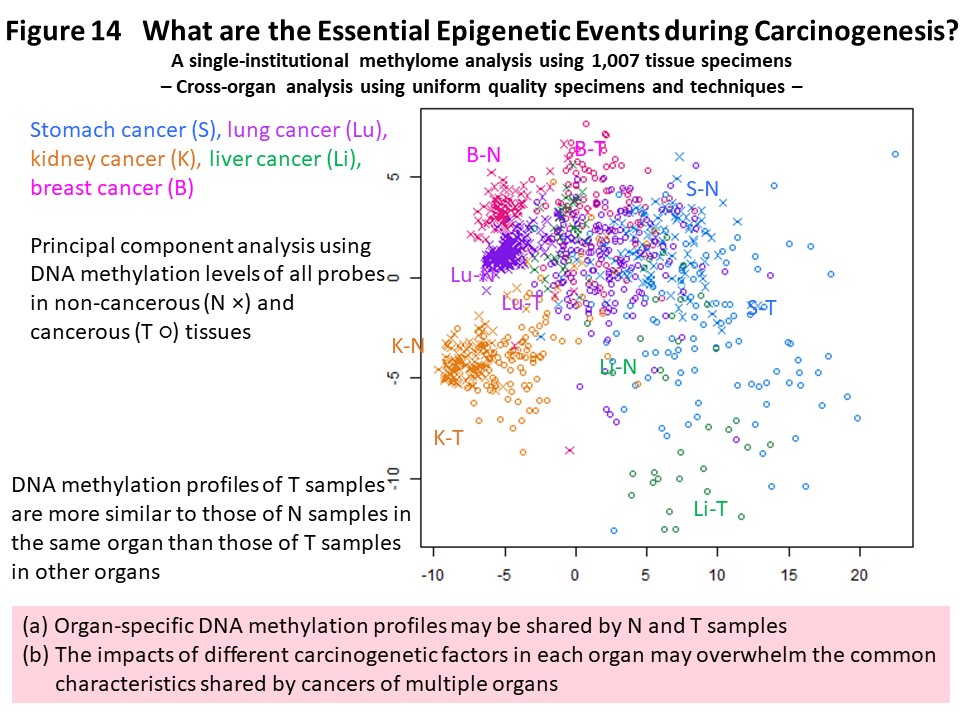
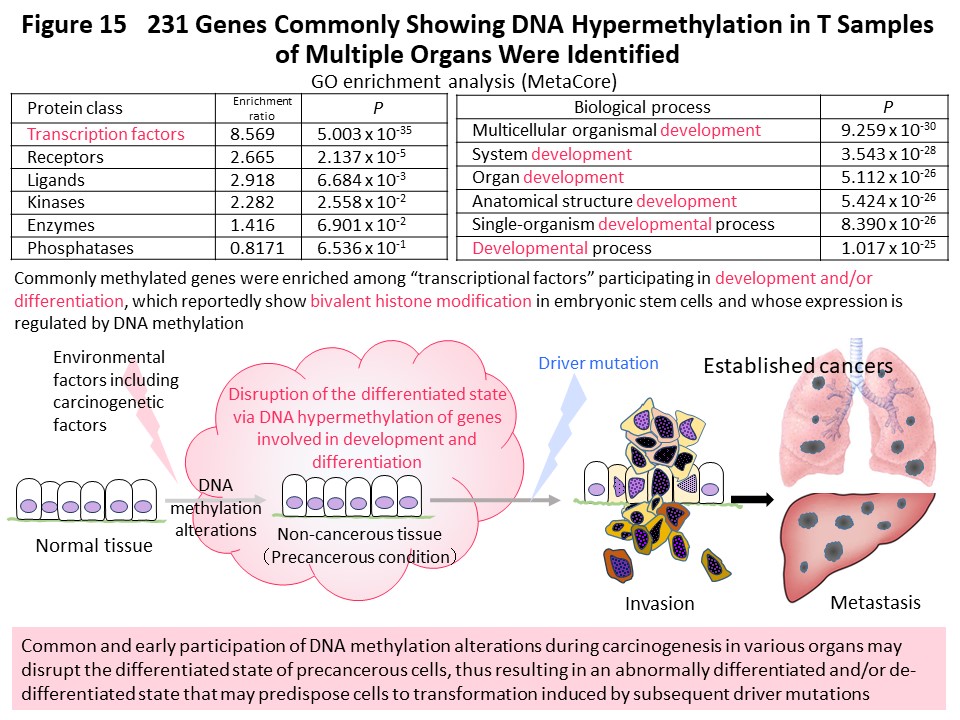
In order to advance understanding of DNA methylation abnormalities that are shared by cancers of various organs, we have used methylome data from more than 1000 samples collected and analyzed ourselves using unique technology. We compared the data across organs and found that the DNA methylation profile of cancer tissues in one organ was more similar to the DNA methylation profiles of non-cancerous tissues in the same organ than to cancers in other organs (Fig. 14) (Carcinogenesis, 38: 241, 2017). It was suggested that cancer maintains an organ-specific DNA methylation profile, or a DNA methylation profile based on the carcinogenic factors specific to that organ, which has been established in both non-cancerous and cancerous tissues. Many genes that commonly exhibit DNA methylation abnormalities in multiple organ cancers encode transcription factors involved in development and differentiation, the expression levels of which are reportedly regulated by DNA methylation (Fig. 15) (Carcinogenesis, 38: 241, 2017). We think it is important that abnormal DNA methylation of these genes occurs at the precancerous stage, disrupting the differentiation state of cells and tissues, then predisposing cells that are the origin of cancer to accept genetic abnormalities referred to as driver mutations.
Since we first reported DNA methylation abnormality in chronic hepatitis or liver cirrhosis, which is a precancerous condition due to hepatitis virus infection (Jpn J Cancer Res, 87: 1210, 1996), we have focused especially on the significance of DNA methylation abnormalities in multistage hepatocarcinogenesis. During the development of viral hepatitis-related hepatocellular carcinoma, DNA methylation abnormalities precede chromosomal instability (Hepatology, 32: 970, 2000) and are associated with DNA methyltransferase expression and/or splicing abnormalities (Int J Cancer, 105: 527, 2003; Proc Natl Acad Sci USA, 99: 10060, 2002). Criteria for carcinogenetic risk estimation (Int J Cancer, 129: 1170, 2011) and prognostication (Int J Cancer, 125: 2854, 2009) have been developed for viral hepatitis-related hepatocellular carcinoma.
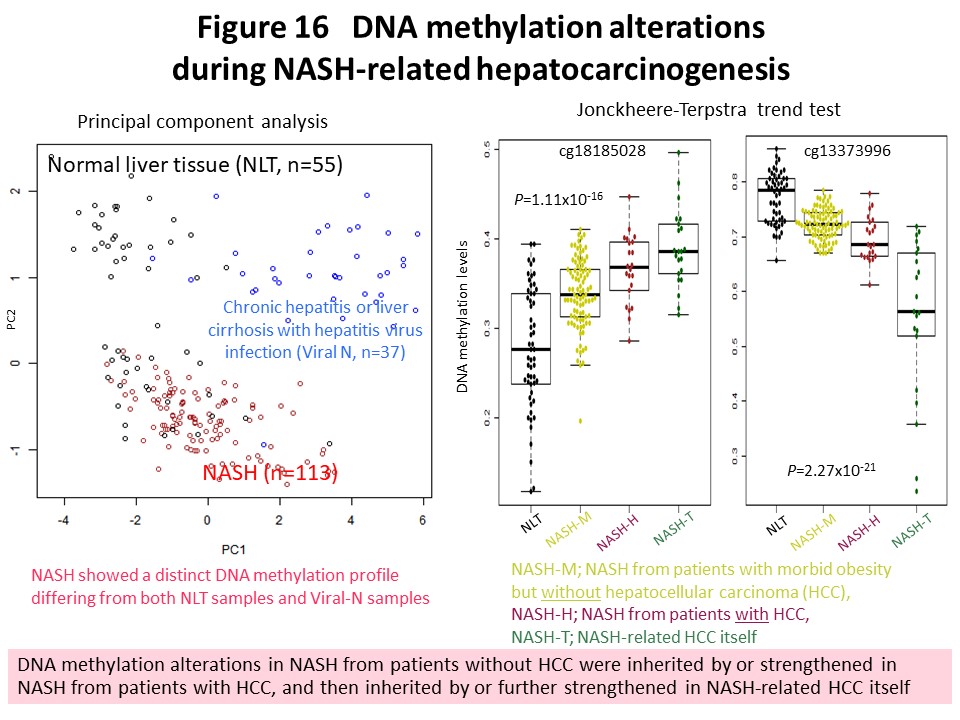
Recently we have been focusing on non-alcoholic steatohepatitis (NASH)-derived hepatocellular carcinoma, the incidence of which has been increasing rapidly in recent years. We found an established NASH-specific DNA methylation profile that was different from such profiles in normal liver tissue and liver tissue showing chronic hepatitis or cirrhosis due to hepatitis virus infection (Carcinogenesis, 38: 261, 2017). NASH-specific DNA methylation abnormality gradually increases from NASH that has not yet developed into hepatocellular carcinoma to NASH that has already done so, and is further inherited by or strengthened in the NASH-derived hepatocellular carcinoma itself (Fig. 16). The histone methyltransferase WHSC1, which shows NASH-specific DNA methylation abnormality, is considered a candidate therapeutic target for NASH-derived hepatocellular carcinoma (Carcinogenesis, 38: 261, 2017). We have further identified cancer-related genes in NASH-derived hepatocellular carcinoma that may result in stable gene expression alteration due to abnormal DNA methylation. Furthermore, clinical realization of cancer risk diagnosis in patients with NASH is considered to be a desirable medical goal, and we are working vigorously with the aim of achieving this.
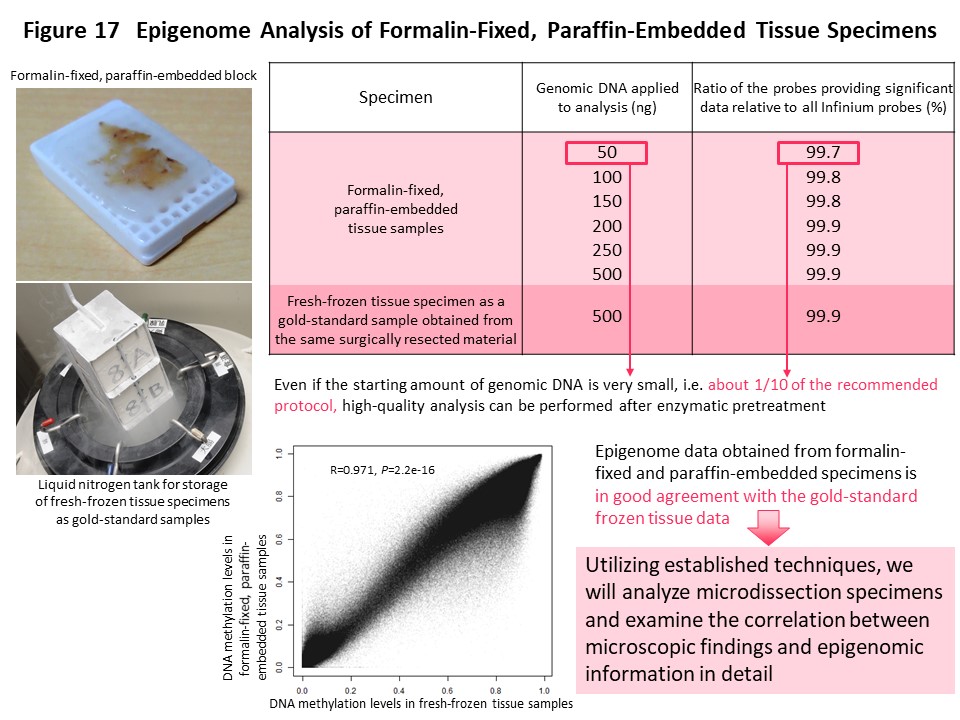
Epigenome information is more plastic than genome information, and DNA methylation abnormalities are affected to various degrees by the diversity of the microenvironment around cancer cells. Therefore, histological heterogeneity evident in cancers during routine microscopy work for pathological diagnosis may be explainable in terms of DNA methylation abnormalities rather than gene mutations. In order to more precisely correlate abnormal DNA methylation with histological heterogeneity of cancer, it is necessary to perform genome-wide DNA methylation analysis in an area showing various degrees of cellular and structural atypia or various degrees of cancer-stromal interaction. Microdissected specimens are tiny tissue fragments that have been cut out from formalin-fixed paraffin-embedded tissue using a laser under microscopic observation. It is difficult to perform such analysis on microdissectied specimens because genomic DNA is denatured during formalin fixation and paraffin embedding. We therefore repaired the genomic DNA enzymatically to facilitate genome-wide DNA methylation analysis with even a small amount of DNA extracted from microdissected samples (Fig. 17) (Pathol Int, 68: 633, 2018).
Since the establishment of this analytical technique, it has been applied to urothelial carcinoma, which shows a variety of histological features such as superficial papillary carcinoma, widely spreading carcinomas in situ, and nodular invasive carcinoma within any given tumor from a single patient. Microdissected specimens have also been collected from surgically resected specimens of glioblastoma, in which tumor blood vessel density is varied and necrotic lesions are scattered within a single tumor. In addition, such specimens have been collected from multiple areas of a single renal cell carcinoma showing different nuclear grades. In renal cell carcinoma, we have conducted multilayer/integrated omics analysis including the genome, epigenome, transcriptome, proteome and metabolome. Currently, glycoproteome analysis is being performed using microdissected specimens of renal cell carcinoma, as a new omics layer, to further identify molecules determining the biological characteristics of cancer.
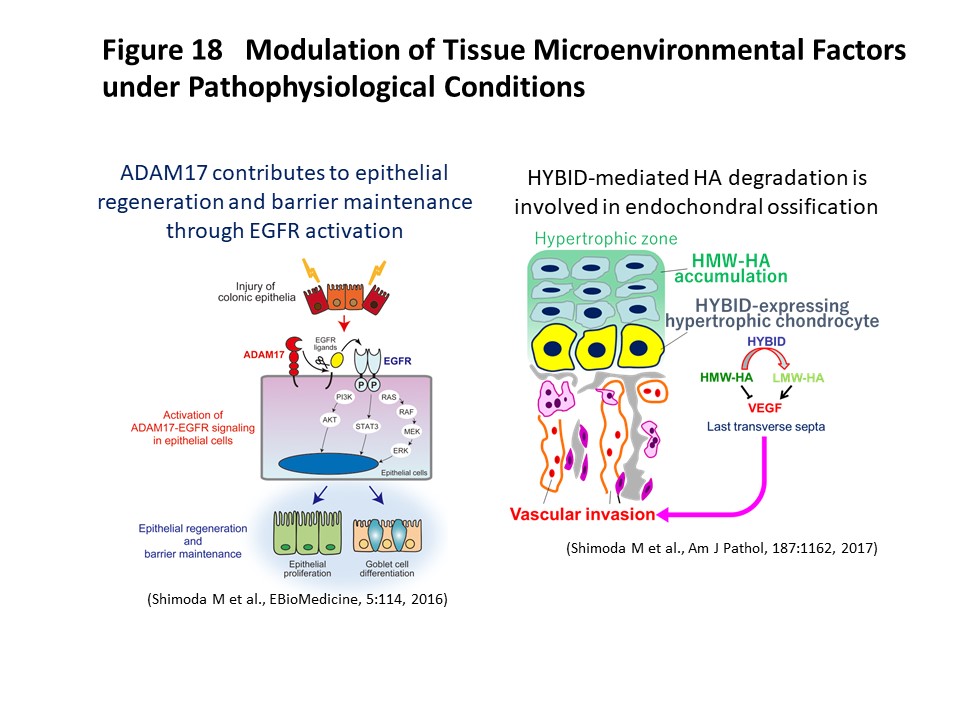
Tissue microenvironmental factors include extracellular matrix molecules and various bioactive factors. They are known to play a crucial role in maintenance of tissue homeostasis, and dysregulation of these factors is involved in tissue destruction and contributes to the pathogenesis of various diseases. We have investigated the significance of tissue microenvironmental factors modulated by metalloproteinases such as ADAMs (a disintegrin and metalloproteinases) and MMPs (matrix metalloproteinases) during tumor progression. In human lung and breast carcinomas, we have shown that ADAM28 is overexpressed predominantly by carcinoma cells and that its expression correlates with lymph node metastasis (Int J Cancer, 118:263, 2006). Mechanistically, ADAM28 promotes cell proliferation by reactivation of insulin-like growth factor-1 through digestion of IGF-binding protein-3 and also enhances tumor metastasis by promoting P-selectin glycoprotein ligand-1/P-selectin-mediated tumor cell rolling adhesion to endothelial cells and escape from von Willebrand factor-induced apoptosis (Cancer Res, 66:9913, 2006; J Biol Chem, 282:25864, 2007; J Natl Cancer Inst, 104:906, 2012). We have further developed a human antibody against ADAM28 that can inhibit its activity in vivo, suggesting that this neutralizing antibody would be useful for treatment of cancer patients in the future. Dysregulation of tissue microenvironmental factors is also observed in non-cancerous diseases and we have also investigated their roles under pathophysiological conditions such as aortic dissection (Atherosclerosis, 218:470, 2011; Circulation, 126:3070, 2012; Circ Res, 116:612, 2015), ulcerative colitis (EBioMedicine, 5:114, 2016), osteoarthritis (Am J Pathol, 188:2109, 2018), muscle regeneration (J Biol Chem, 290:28456, 2015) and endochondral ossification (Am J Pathol, 187:1162, 2017) (Figure 18). We are further investigating the mechanisms of tissue microenvironment establishment under various pathophysiological conditions, focusing especially on tumor development and gastrointestinal diseases.
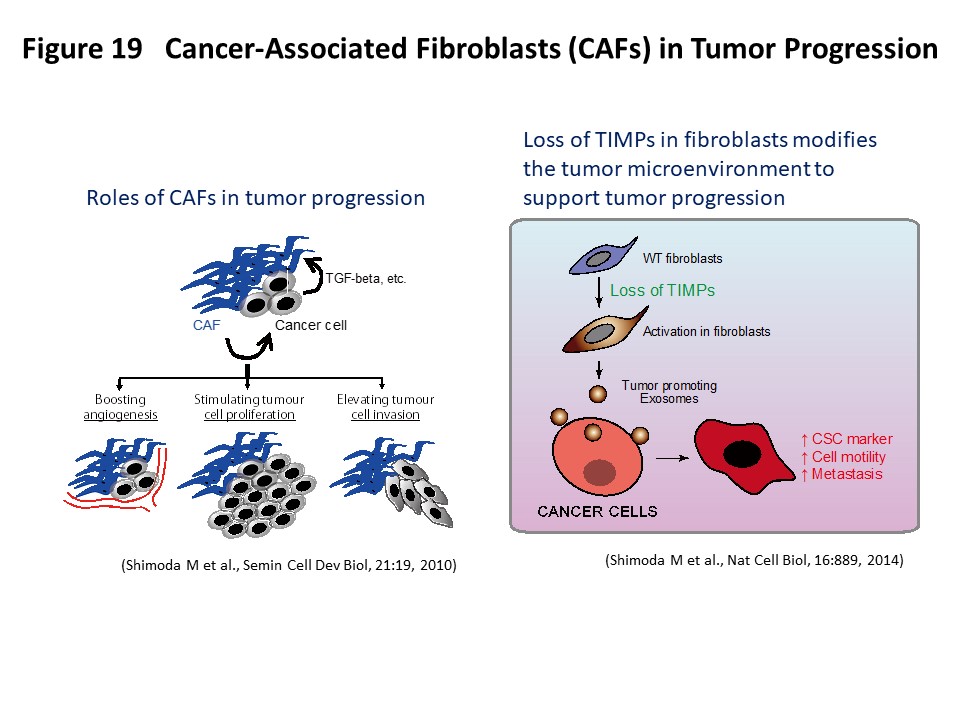
Tumors are complex tissues composed of carcinoma cells and a surrounding stroma, which includes various different types of mesenchymal cells and extracellular matrix molecules. Carcinoma-associated fibroblasts (CAFs) are frequently observed in the stroma of human carcinomas and their presence in large numbers is often associated with the development of high-grade malignancies and poor prognoses (Semin Cell Dev Biol, 21:19, 2010). CAFs are known to acquire an activated phenotype and have been shown to stimulate tumor cell proliferation, promote tumor cell invasion and boost angiogenesis by secreting various growth factors and cytokines. Although it is conceivable to therapeutically target CAFs to elicit anti-tumor responses, CAFs are also thought to have multiple origins and comprise a heterogeneous population of cells (J Exp Med, 211:1503, 2014). Therefore, it is important to understand the unique roles of the different subpopulations of CAFs. We have recently generated quadruple Timp (tissue inhibitor of metalloproteinases)-deficient fibroblasts to elicit metalloproteinase activity within the tumor–stromal compartment and demonstrated that complete loss of TIMPs allows the acquisition of hallmarks of CAF function. Detailed analysis has revealed that increased metalloproteinase activity is involved in the induction of this activated fibroblast state and that fibroblast-derived exosomes act as a tumor-promoting vector for stroma–cancer communication (Nat Cell Biol, 16:889, 2014; Biochim Biophys Acta Mol Cell Res, 1864:1989, 2017) (Figure 19). We are further evaluating various CAF cell lines for their clinicopathological and biological properties as well as gene expression patterns to functionally characterize active CAFs. In this research project, we hope to contribute to the classification and precise molecular definition of fibroblast subtypes in the tumor stroma, which may offer additional insights into CAF biology and suggest clinical opportunities for new cancer therapies.